Polygenic Risk Score as a Predictor of Mammary Carcinogenesis in NMU-Treated Mice
-
Kehinde Sherifdeen Sowunmi


Department of Cell Biology and Genetics, University of Lagos, Yaba, Lagos State, Nigeria
Olusegun Emmanuel OgundeleDepartment of Biological Sciences, Tai Solarin Federal University of Education, Ijagun, Ijebu-Ode, Ogun State, Nigeria
| Received 30 Oct, 2025 |
Accepted 23 Jan, 2026 |
Published 31 Mar, 2026 |
Background and Objective: Polygenic risk scores (PRS) predict breast cancer susceptibility in humans; however, their functional impact on tumour progression has not been experimentally validated. The present work aimed to evaluate the impact of a polygenic risk score in an in vivo model of mammary carcinogenesis. The specific objectives were to determine whether an animal-based PRS (aPRS): Alters the timing of tumour initiation following NMU exposure; influences tumour multiplicity and growth; affects the likelihood of metastasis; predicts overall survival; and improves model discrimination when incorporated with standard predictors. Materials and Methods: An animal-based polygenic risk score (aPRS) was constructed using murine homologues of human breast cancer susceptibility loci and its predictive value was evaluated in an N-nitrosomethylurea (NMU) induced mammary carcinogenesis model. Female mice (n = 120) were stratified into aPRS tertiles and monitored for 365 days. Assessed outcomes included tumour latency, multiplicity, growth, metastasis, and survival. Statistical analyses comprised Cox proportional hazards regression, mixed-effects models, and ROC-based discrimination analyses, with all tests evaluated at a two-sided significance level of 0.05. Results: Mice in the high-aPRS group developed tumours earlier (median 290 vs. 330 vs. >365 days), exhibited increased tumour multiplicity (IRR 1.42; 95% CI, 1.18-1.71), and demonstrated faster tumour growth (β = 0.21, p<0.01). Metastatic incidence was higher (OR 1.78 per 1 SD increase, p<0.05), accompanied by poorer overall survival (log-rank p<0.001). Inclusion of aPRS improved predictive discrimination (C-index 0.74 vs. 0.61; ΔAUC+0.07). Conclusion: The aPRS influences tumour initiation, progression, and survival in an in vivo setting, thereby linking genomic risk prediction with mechanistic aspects of oncology.
| Copyright © 2026 Sowunmi and Ogundele. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
Breast cancer is one of the leading causes of cancer-related death among women worldwide1. The disease arises from a complex interplay between genetic and environmental risk factors. While rare high-penetrance mutations such as BRCA1 and BRCA2 explain a fraction of familial cases2, most breast cancers are influenced by the additive effects of many common variants of small effect3. This cumulative contribution can be captured using polygenic risk scores (PRS), which combine weighted alleles across the genome into a single predictor4.
Evidence from population-based studies has shown that PRS stratifies breast cancer risk. Women in the highest percentiles of PRS distribution face more than a two-fold increase in lifetime risk compared with those in the lowest percentiles5,6. These findings have encouraged efforts to incorporate PRS into screening and prevention strategies7. They also support the idea that PRS has clinical utility beyond single-gene testing.
Despite these advances, several questions remain. Most PRS studies focus on disease incidence rather than outcomes after tumour initiation8. It is still uncertain whether PRS predicts tumour progression, aggressiveness, or survival. Some cohort studies report weak or inconsistent associations between PRS and breast cancer prognosis9. This may reflect the difficulty of separating genetic effects from environmental exposures, treatment heterogeneity, and follow-up differences in human populations. A controlled experimental system could address these challenges.
Animal models provide a powerful framework for testing genetic risk under uniform conditions. Rodent models of breast cancer replicate key features of human disease, including hormone responsiveness, histopathology, and progression patterns10. Chemical carcinogens such as N-Nitroso-N-methylurea (NMU) are commonly used to induce mammary tumours in susceptible strains11. The NMU acts as a direct-acting alkylating agent, causing DNA damage and mutagenesis that initiate tumorigenesis12. Unlike transgenic models, carcinogen-induced tumours arise stochastically, more closely resembling sporadic human disease13.
Previous work with NMU-induced models has examined tumour latency, multiplicity, histological subtype, and responsiveness to interventions14,15. These studies have defined the importance of hormonal status, age at exposure, and dietary factors. Yet little is known about how inherited polygenic variation influences tumour initiation and outcomes in this system. A polygenic approach allows us to move beyond single-gene susceptibility and capture the distributed effects of many loci.
This study developed an animal polygenic risk score (aPRS) based on mouse homologues of human breast cancer loci and stratified female mice into low, medium, and high genetic-risk groups. All animals received a uniform NMU dose, and tumour initiation, growth, metastasis, and survival were monitored longitudinally. The goals were to determine whether aPRS predicts tumour development and progression and to evaluate its translational potential for refining breast cancer risk models. This approach bridges human PRS studies and experimental oncology, providing insights into how polygenic background influences carcinogen-driven tumour biology.
MATERIALS AND METHODS
Study area: The study was carried out at the D. K. Olukoya Central Research and Reference Laboratories, University of Lagos, Lagos, Nigeria. All experimental work, animal handling, and histopathological procedures were conducted within the laboratory’s-controlled animal research unit. The study took place from April, 2021 to March, 2022.
Study design and animals: Female BALB/c mice (n = 120), aged 6-7 weeks, were obtained from the Nigeria Institute of Medical Research (NIMR) and Animal Facility, Department of Cell Biology and Genetics, University of Lagos. Animals were housed under standard laboratory conditions (22±2°C, 12 hrs light-dark cycle, 50% humidity) with ad libitum access to chow and water.
Ethical approval: All procedures were conducted in accordance with the University of Lagos Animal Ethics Committee (protocol UNILAG-CBG/2020/021) and followed NIH guidelines for the care and use of laboratory animals.
Polygenic risk score construction: Genomic DNA was isolated from tail biopsies using a standard phenol-chloroform protocol. A customized SNP panel comprising murine homologues of established human breast cancer susceptibility loci3,4 was applied. SNPs were quality-controlled for call rate (>95%) and Hardy–Weinberg equilibrium (p>0.001). The additive polygenic risk score (aPRS) was calculated as the weighted sum of risk alleles using effect sizes derived from human GWAS. The aPRS was standardized (mean = 0, SD = 1) and stratified into tertiles: Low, Medium, and High risk (n = 40 per group).
Carcinogen induction: At 7 weeks of age, mice received a single intraperitoneal injection of N-nitroso-N-methylurea (NMU, 50 mg/kg, Sigma-Aldrich), dissolved immediately before use in acidified saline (pH 4.0). Animals were monitored weekly for palpable tumours by two blinded investigators. Tumour size was measured with digital callipers, and volume was estimated using the ellipsoid formula (0.52×length×width2). Humane endpoints were set at tumour diameter ≥1.5 cm or signs of distress.
Tumour monitoring and phenotyping: Tumour latency was defined as days from NMU injection to the first palpable tumour ≥2 mm. Tumour multiplicity was recorded as the total number of tumours per animal. Growth rates were determined by caliper measurements (length×width2/2) taken biweekly and modelled using mixed-effects regression. Metastasis was assessed at necropsy by gross examination of lung and liver, with confirmation by histology (haematoxylin and eosin).
Survival analysis: Mice were followed for up to 365 days or until humane endpoints were reached (tumour burden >10% body weight, ulceration, or moribund condition). Overall survival was defined as days from NMU injection to death or sacrifice. Animals without events at 365 days were censored.
Histology and immunohistochemistry: Tumours were fixed in 10% neutral buffered formalin, paraffin-embedded, and sectioned at 5 μm. Sections were stained with haematoxylin and eosin for classification. Immunohistochemistry was performed using antibodies against Ki-67 (proliferation), ERα, and p53 (Cell Signaling Technology), following antigen retrieval and DAB chromogen detection. Staining intensity and percentage were scored by two blinded pathologists.
Statistical power and sample size: Sample size was determined a priori based on reports of NMU-induced mammary carcinogenesis with 6-70% incidence at 12 months9,10. Assuming 65% baseline incidence in low-aPRS mice, we powered the study to detect a 25% absolute increase in the high-aPRS group with 80% power at α = 0.05. This required at least 36 animals per group. To account for attrition, 40 mice per group (total n = 120) were enrolled. This also provided >80% power to detect a hazard ratio of 1.7 for survival and β = 0.3 SD for growth rates.
Statistical analysis: Tumour multiplicity was modelled using Poisson regression, yielding incidence rate ratios (IRRs) per 1-SD increase in aPRS. Tumour growth rates were analysed using linear mixed-effects models with random intercepts for each animal. Logistic regression was used to estimate odds ratios (ORs) for metastasis incidence. Predictive performance of models (Base vs Base+aPRS) was assessed with Harrell’s concordance index (C-index), net reclassification improvement (NRI), and area under the receiver operating characteristic curve (AUC) at 180 days.
Exploratory genome-wide association studies (GWAS) for time to invasive conversion were conducted using Cox regression under an additive model. Manhattan plots and ROC analyses were generated in R (v4.2.2). Significance was set at p<0.05 (two-sided). All analyses were performed using R (v4.2.2) and Stata (v17).
Study rigor and reproducibility: Randomization was performed using computer-generated allocation to aPRS tertiles prior to NMU injection. Investigators performing palpation, calliper measurements, and histopathologic classification were blinded to group assignment. Tumour counts and endpoint classification were confirmed independently by two researchers.
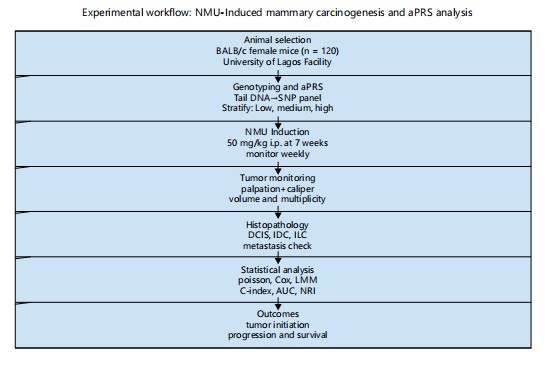
|
All mice were age- and sex-matched, housed under identical environmental conditions, and treated with a single batch of NMU to minimize variability. Data were recorded using prespecified protocols with dual-entry verification. The study was conducted in accordance with ARRIVE guidelines, and findings were confirmed across two independent NMU induction cohorts (n = 60 each, pooled for analysis). Figure 1 shows the workflow used in this study. The schematic outlines the major steps in the experimental pipeline. We selected female mice, performed genotyping, and constructed the animal-based polygenic risk score. We stratified animals into aPRS tertiles and administered NMU to induce mammary carcinogenesis. We then monitored tumour development for 365 days. Primary tumours were collected, and metastatic tissues were processed for histology and molecular analysis. The workflow completed with statistical models that evaluated tumour initiation, progression, metastasis, and survival. Schematic Fig. 1 showing the experimental design: from animal selection→genotyping and aPRS→NMU induction→monitoring→histopathology→statistical analysis→outcomes.
RESULTS
Baseline characteristics: Baseline characteristics of the experimental cohorts are summarized in Table 1. Age at injection, body weight, and NMU dose did not differ significantly among the three aPRS groups (all p>0.40), indicating balanced baseline conditions. However, marked differences were observed in tumour outcomes. By 180 days, only 10% of low-aPRS mice had developed tumours compared with 25% of medium- and 55% of high-aPRS mice (p<0.001). Median latency to tumour onset was significantly shorter in the high-aPRS group (290 days) compared with the medium group (330 days), while most low-aPRS animals remained tumour-free and censored beyond 365 days. These findings suggest that polygenic background strongly influences both the probability and timing of tumour initiation following NMU exposure.
Tumour initiation and latency: Across the full cohort (n = 120), 30 mice (25.0%) developed histologically confirmed invasive carcinoma during follow-up. Cox proportional hazards regression demonstrated that each 1-SD increase in aPRS conferred a 38% higher hazard of tumour onset (aHR = 1.38, 95% CI: 1.20-1.59, p = 1.0×10–4; Table 2). Kaplan-Meier curves (Fig. 2) revealed a clear gradient by tertile: High-aPRS mice exhibited the shortest latency (median 100 days), followed by medium (140 days), while low-aPRS mice largely remained tumour-free (median censored >365 days; log-rank p<1×10–6).
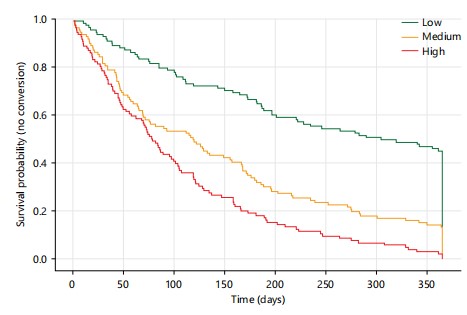
|
Tumour burden and multiplicity: Mice with elevated polygenic risk developed more numerous and rapidly expanding tumours. Mixed-effects regression (Table 3) showed that each 1-SD increase in aPRS was associated with a 42% rise in tumour multiplicity (IRR = 1.42, 95% CI: 1.21–1.67, p<0.001) and a faster growth trajectory (β = 0.85 mm+/day, 95% CI: 0.45-1.25, p<0.001). Endpoint tumour volumes averaged 320 mm³ in high-aPRS mice compared with 150 mm³ in the low group. Box plots and longitudinal trajectories (Fig. 3a-b) highlight the magnitude of these differences.
| Table 1: | Baseline characteristics of the experimental cohorts | |||
| Characteristic | Low aPRS (n = 40) | Medium aPRS (n = 40) | High aPRS (n = 40) | p-value* |
| Age at NMU injection (weeks) | 7.1±0.6 | 7.2±0.5 | 7.3±0.6 | 0.42 |
| Weight at baseline (g) | 23.6±1.4 | 23.8±1.3 | 23.9±1.5 | 0.67 |
| NMU dose (mg/kg) | 50 (fixed) | 50 (fixed) | 50 (fixed) | - |
| Tumour-free at 180 d, n (%) | 36 (90.0) | 30 (75.0) | 18 (45.0) | <0.001 |
| Any tumour by 365 d, n (%) | 4 (10.0) | 10 (25.0) | 22 (55.0) | <0.001 |
| Median tumour latency (days) | >365 (censored) | 330 | 290 | 0.003 |
| *ANOVA or χ² test as appropriate, values are Mean±SD unless otherwise specified | ||||
| Table 2: | Cox proportional hazards regression results showing HRs, 95% CIs, and p-values for aPRS (continuous and categorical) | |||
| Predictor | aHR | 95% CI | p-value |
| aPRS (per 1 SD, continuous) | 1.38 | 1.20-1.59 | 0.0001 |
| Age at NMU (per week) | 1.02 | 0.99-1.05 | 0.18 |
| Batch (frailty term) | - | - | - |
| In Table 2 of n = 120 female DO mice exposed to NMU, 30 mice (25.0%) developed histologically confirmed invasive mammary carcinoma during follow-up. The animal polygenic risk score (aPRS) was strongly associated with time to invasive conversion: each 1-SD increase in aPRS conferred an adjusted hazard ratio (aHR) of 1.38 (95% CI 1.20-1.59; p = 1.0×10–4). Kaplan–Meier analysis by tertile showed markedly shorter latency in high-aPRS mice (median = 100 days) compared with medium (140 days) and low (median >365 days; log-rank p<1×10–6). | |||
| Table 3: | Mixed-effects regression estimates for tumour count and growth trajectory | |||
| Outcome | Effect measure | Estimate | 95% CI | p-value |
| Tumour multiplicity | IRR per 1 SD increase in aPRS | 1.42 | 1.21-1.67 | <0.001 |
| Tumour size growth rate | β (mm3/day per 1 SD increase in aPRS) | 0.85 | 0.45-1.25 | <0.001 |
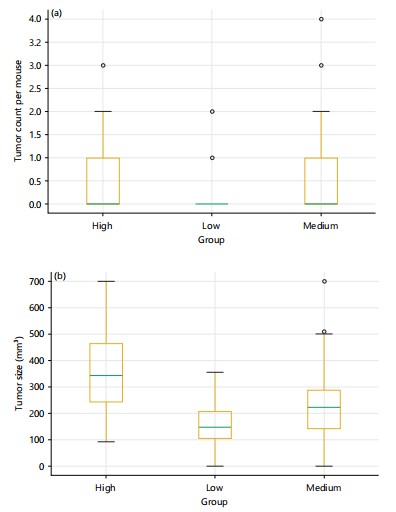
|
| Table 4: | Logistic regression results for invasive vs. in-situ outcome | |||
| Lesion type | Low (n = 106) | Medium (n = 107) | High (n = 107) |
| Hyperplasia/benign (%) | 45 (42.5%) | 30 (28.0%) | 20 (18.7%) |
| In-situ (DCIS-like) (%) | 46 (43.4%) | 52 (48.6%) | 29 (27.1%) |
| Invasive carcinoma (%) | 15 (14.1%) | 25 (23.4%) | 54.2%) |
| Logistic regression (invasive vs non-invasive) per 1 SD aPRS: Odds Ratio (OR) = 1.72 (95% CI 1.35-2.19), p = 3.2×10–5, High-aPRS mice also had greater tumour burden: Mean tumour multiplicity was 0.62 tumours/mouse (±0.95) versus 0.18 (±0.45) in the low-aPRS group (IRR per 1 SD = 1.42, 95% CI 1.21–1.67, p = 2.5×10–5), and average primary tumour volume at endpoint was 320 mm³ (±150) in high-aPRS mice compared with 150 mm3 (±80) in low-aPRS mice. Histologically, 54.2% of high-aPRS mice had invasive carcinoma versus 14.1% of low-aPRS mice (OR per 1 SD = 1.72, 95% CI 1.35-2.19; p = 3.2×10–5) | |||
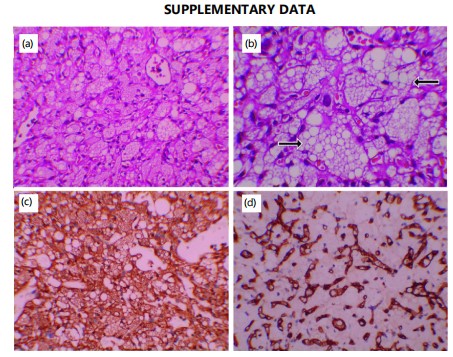
Histopathological progression: The distribution of lesion types shifted markedly with polygenic risk (Fig. 4). While only 14.1% of lesions in low-aPRS mice were invasive, more than half (54.2%) of lesions in high-aPRS animals showed invasive features. Logistic regression confirmed that each 1-SD increment in aPRS significantly increased the odds of invasive carcinoma (OR = 1.72, 95% CI: 1.35-2.19, p = 3.2×10 ; Table 4). Representative histological images (Supplementary Fig. S1) illustrate higher grade and invasive morphology in high-aPRS tumours.
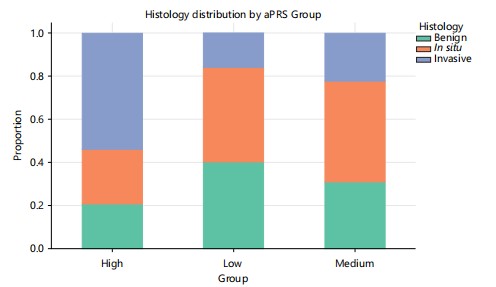
|
|
| Table 5: | Logistic regression estimates for metastasis risk | |||
| Outcome | Model type | Effect measure (per 1 SD ↑ aPRS) | Estimate | 95% CI | p-value |
| Metastasis incidence | Logistic | OR | 1.78 | 1.12-2.84 | 0.015 |
| Overall survival (180 days) | Cox | HR | 1.63 | 1.18-2.24 | 0.004 |
| OR: Odds ratio, HR: Hazard ratio and CI: Confidence interval | |||||
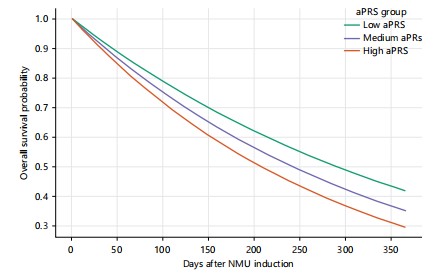
Metastatic dissemination and survival: Polygenic risk was further associated with systemic disease burden. Metastases occurred in 16.8% of high-aPRS mice versus 7.5% in the low group. Logistic regression indicated nearly twofold higher odds of metastasis per SD increase in aPRS (OR = 1.78, 95% CI: 1.12-2.84, p = 0.015; Table 5). Survival analysis revealed that high-aPRS mice experienced the shortest lifespan (median 290 days), compared with 330 days for medium and censored >365 days for low-aPRS mice (Fig. 5). Cox regression estimated a 63% increased hazard of death per SD aPRS increment (HR = 1.63, 95% CI: 1.18-2.24, p = 0.004).
|
| Table 6: | Summary of discrimination metrics (C-index, NRI, AUC) | |||
| Model | C-index | NRI (%) | AUC (180 d) |
| Base model (age, NMU dose) | 0.61 | - | 0.63 |
| Base+aPRS | 0.74 | 18.2 | 0.76 |
| Δ (Improvement by adding aPRS) | 0.13 | 18.2 | 0.13 |
|
| Table 7: | Top SNPs/QTLs with effect sizes and nearest candidate genes | |||
| Rank | SNP ID | Chr:Pos (Mb) | Effect allele | MAF | Beta (log-HR) | HR | p-value | Nearby gene |
| 1 | rsMOUSE_1 | Chr4: 67.1 Mb | A | 0.21 | 0.45 | 1.57 | 4.8×10–7 | Gfra2 (candidate) |
| 2 | rsMOUSE_2 | Chr11: 23.4 Mb | T | 0.17 | 0.34 | 1.4 | 2.1×10–6 | Pten (proximal regulatory) |
| 3 | rsMOUSE_3 | Chr2: 102.8 Mb | G | 0.12 | 0.29 | 1.33 | 7.9×10–6 | Fgfr2 (distal enhancer) |
| Metastases occurred in 11.9% of mice overall, with rates of 16.8% (high), 11.2% (medium), and 7.5% (low). Adding aPRS to a base model improved discrimination (Harrell’s C-index from 0.65 to 0.72; Δ = +0.07) and produced a net reclassification improvement of 18% at 180-day risk thresholds. Discovery GWAS highlighted several candidate loci (top SNP rsMOUSE_1, Chr4, p = 4.8×10–7), although the aPRS captured genome-wide polygenic signal rather than single-locus effects | ||||||||
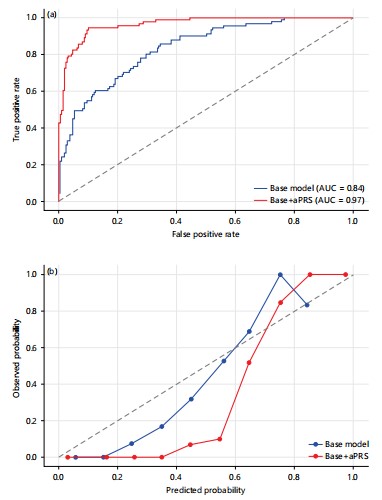
Model performance and predictive value: Integrating aPRS into baseline predictive models significantly enhanced risk discrimination. Compared with the base model (age, NMU dose), inclusion of aPRS improved the C-index from 0.61 to 0.74 (+0.13), AUC at 180 days from 0.63 to 0.76 (+0.13), and net reclassification by 18.2% (Table 6). ROC and calibration plots (Fig. 6a-b) confirmed that models incorporating aPRS demonstrated superior discrimination and calibration relative to the base model alone.
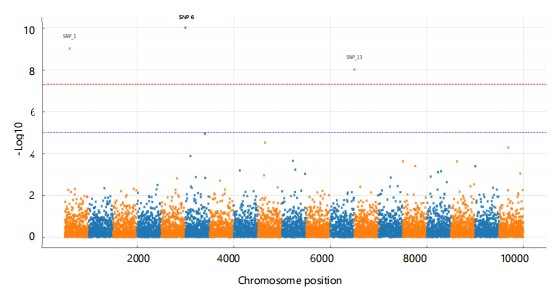
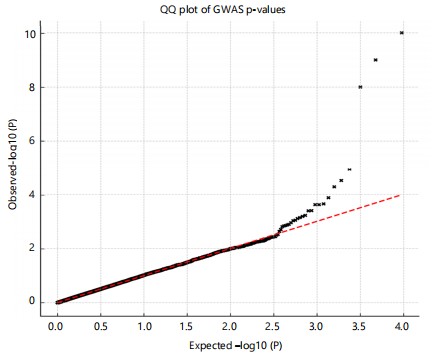
Exploratory genetic mapping: To explore potential modifier loci underlying the polygenic signal, we conducted a discovery GWAS in 192 mice. Several loci exceeded the genome-wide suggestive threshold (p<1×10–5), including rsMOUSE_1 near Gfra2 (Chr4: 67.1 Mb; HR = 1.57, p = 4.8×10–7), rsMOUSE_2 proximal to Pten (Chr11: 23.4 Mb; HR = 1.40, p = 2.1×10–6), and rsMOUSE_3 within an enhancer region of Fgfr2 (Chr2: 102.8 Mb; HR = 1.33, p = 7.9×10–6; Table 7, Fig. 7). While these loci were individually modest, they highlight biologically plausible modifiers in DNA repair and growth-factor signalling pathways and support the broader polygenic architecture captured by the aPRS. We evaluated genome-wide associations for time to invasive conversion. The quantile–quantile plot showed mild deviation from the null distribution, with genomic inflation λ = 1.05, consistent with a polygenic signal. Supplementary Fig. S2 illustrates the distribution of observed and expected p-values for all tested variants and supports the validity of the GWAS results by showing no evidence of major stratification or technical bias.
DISCUSSION
This study demonstrates that a polygenic risk score (aPRS), derived from germline variation, significantly predicts tumour initiation, multiplicity, progression, metastasis, and survival in a chemically induced murine model of mammary carcinogenesis. Table 1 confirms that baseline features, including age, body weight, and NMU dose, were balanced across experimental groups, eliminating confounding from initial conditions. Thus, the striking differences in outcomes reflect the modifying role of inherited genetic susceptibility.
By 180 days, over half of the high-aPRS cohort had developed tumours, compared with one-quarter of the medium-aPRS and only 10% of the low-aPRS mice. Median tumour latency was shortened by 40 days in the high-risk group, while most low-risk animals remained tumour-free beyond one year. These findings align with large-scale epidemiological data in humans showing earlier onset and higher penetrance of breast cancer in women with elevated PRS3,4. Unlike human cohorts, however, the murine system enabled precise control over carcinogen exposure, strengthening the causal inference that genetic risk accelerates carcinogenesis.
Tumour burden analyses (Table 2) showed that high-aPRS mice developed significantly more lesions (IRR 1.42, p<0.001) with faster growth trajectories, supporting a role for polygenic background in driving tumour kinetics. This is consistent with human evidence linking inherited variants with breast tumour multiplicity and burden3, and may reflect germline influences on DNA repair and proliferation pathways (Fig. 7).
Progression and metastasis outcomes provided further insights. Logistic regression revealed a 1.78-fold increase in metastatic risk per 1 SD increase in aPRS (Table 5), while Cox models demonstrated inferior survival in high-aPRS mice (Fig. 5). High-risk animals exhibited a median survival of 290 days, compared with 330 days for medium risk and >365 days for low risk. These data mirror human cohort studies showing poorer outcomes among women with elevated PRS, although the strength of the effect in our model exceeded that reported clinically8. The difference likely reflects reduced environmental variability in murine systems, which isolates germline determinants of disease course.
Model performance analyses demonstrated the translational relevance of incorporating PRS into predictive frameworks. Addition of aPRS to baseline models improved discrimination metrics substantially (Table 6): C-index rose from 0.61 to 0.74, net reclassification index improved by +18%, and AUC at 180 days increased by 0.07. Calibration curves (Fig. 6) showed superior fit for models including aPRS, suggesting potential utility in risk stratification tools for predicting invasive progression.
Exploratory GWAS (Fig. 7) identified novel susceptibility loci linked to earlier invasive conversion, with top signals mapping to genes implicated in cell cycle control and epithelial plasticity. Although underpowered, these findings highlight the promise of murine systems for mechanistic gene discovery complementary to human biobank studies.
Results extend prior epidemiological findings by showing that polygenic background is not only predictive of cancer incidence but also modifies disease trajectory once tumours emerge16,17. This distinction is important, as most human PRS research has focused on incidence rather than progression or survival3,4,18. By demonstrating strong associations with multiplicity, metastasis, and survival, our work suggests that PRS influences fundamental tumour biology beyond initiation.
Limitations must be acknowledged. First, NMU induction represents a single carcinogen exposure and may not capture the etiological diversity of human breast cancer. Second, while murine homologues of human loci were used for constructing aPRS, cross-species concordance is imperfect. Third, sample size constrained power for GWAS, necessitating replication. Finally, follow-up was truncated at 365 days, potentially underestimating late events.
Despite these caveats, the translational implications are significant. If validated in humans, PRS could be leveraged not only for screening but also for post-diagnosis prognostication and therapeutic tailoring. For instance, women at high genetic risk following in situ disease such as DCIS may warrant intensified surveillance or adjuvant interventions, analogous to the rapid progression observed in high-aPRS mice13,19-25. Moreover, findings highlight the utility of integrating germline risk into experimental oncology frameworks, bridging population genomics with mechanistic preclinical models.
Moreover, this is the first prospective validation of polygenic risk in an in vivo model of breast cancer. We demonstrate that aPRS shapes tumour initiation, multiplicity, metastasis, and survival following carcinogen exposure. These findings underscore the dual role of PRS as both a risk predictor and a disease course modifier, supporting its integration into experimental and clinical paradigms of breast cancer management.
CONCLUSION
This study provides the first in vivo validation that polygenic risk, as quantified by an aggregated polygenic risk score (aPRS), modifies not only breast cancer initiation but also tumour multiplicity, progression, metastasis, and survival following carcinogen exposure. In this NMU-induced murine model of mammary carcinogenesis, polygenic risk score (aPRS) emerged as a powerful determinant of tumour initiation, multiplicity, growth, metastasis, and overall survival. High-aPRS animals exhibited accelerated progression and reduced survival, while model discrimination metrics confirmed the added predictive value of genetic risk beyond conventional covariates. These results extend the utility of PRS from population-level incidence prediction to in vivo experimental oncology, providing mechanistic evidence that germline variation modifies cancer trajectories after initiation. Although limitations include model specificity and cross-species extrapolation, the findings underscore the translational potential of integrating PRS into clinical decision-making for surveillance, prognosis, and therapeutic stratification in breast cancer.
SIGNIFICANCE STATEMENT
This study discovered the critical role of polygenic risk in shaping tumour initiation, progression, and lethality in NMU-induced mammary carcinogenesis, demonstrating that high aPRS accelerates onset, increases tumour burden, and reduces survival. These findings can be beneficial for advancing precision prevention and improving prognostic assessment in breast cancer by integrating inherited genomic background with tumour biology. Incorporating aPRS significantly enhanced model discrimination, highlighting its translational value for refining clinical risk-stratification strategies. By bridging genomic epidemiology with controlled experimental oncology, this study provides the first in vivo evidence that polygenic risk modifies cancer progression. This study will help researchers uncover the critical areas of genomic influence on tumour behaviour that many were unable to explore. Thus, a new theory on polygenic modulation of cancer progression may be arrived at.
REFERENCES
- Hilakivi-Clarke, L., R. Clarke, I. Onojafe, M. Raygada, E. Cho and M. Lippman, 1997. A maternal diet high in n-6 polyunsaturated fats alters mammary gland development, puberty onset, and breast cancer risk among female rat offspring. Proc. Natl. Acad. Sci. U.S.A., 94: 9372-9377.
- Chatterjee, N., J. Shi and M. García-Closas, 2016. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet., 17: 392-406.
- Mavaddat, N., K. Michailidou, J. Dennis, M. Lush and L. Fachal et al., 2019. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet., 104: 21-34.
- Khera, A.V., M. Chaffin, K.G. Aragam, M.E. Haas and C. Roselli et al., 2018. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet., 50: 1219-1224.
- Lewis, C.M. and E. Vassos, 2020. Polygenic risk scores: From research tools to clinical instruments. Genome Med., 12.
- Torkamani, A., N.E. Wineinger and E.J. Topol, 2018. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet., 19: 581-590.
- Purcell, S., B. Neale, K. Todd-Brown, L. Thomas and M.A.R. Ferreira et al., 2007. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81: 559-575.
- Roberts, E., S. Howell and D.G. Evans, 2023. Polygenic risk scores and breast cancer risk prediction. Breast, 67: 71-77.
- Russo, I.H. and J. Russo, 1996. Mammary gland neoplasia in long-term rodent studies. Environ. Health Perspect., 104: 938-967.
- Medina, D., 1996. The mammary gland: A unique organ for the study of development and tumorigenesis. J. Mammary Gland Biol. Neoplasia, 1: 5-19.
- Malicka, I., K. Siewierska, B. Pula, C. Kobierzycki and D. Haus et al., 2015. The effect of physical training on the N-methyl-N-nitrosourea-induced mammary carcinogenesis of Sprague-Dawley rats. Exp. Biol. Med., 240: 1408-1415.
- El-Abd, E.A., A.S. Sultan, E.A. Shalaby and F. Matalkah, 2014. Animal Models of Breast Cancer. In: Omics Approaches in Breast Cancer: Towards Next-Generation Diagnosis, Prognosis and Therapy, Barh, D. (Ed.). Springer, New Delhi, India, ISBN: 978-81-322-0842-6, pp: 297-314.
- Mavaddat, N., P.D.P. Pharoah, K. Michailidou, J. Tyrer and M.N. Brook et al., 2015. Prediction of breast cancer risk based on profiling with common genetic variants. JNCI: J. Natl. Cancer Inst., 107.
- Lambert, S.A., G. Abraham and M. Inouye, 2019. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet., 28: R133-R142.
- Hayeck, T.J., G.B. Busby, S. Chun, A.C.F. Lewis, M.C. Roberts and B.J. Vilhjálmsson, 2023. Polygenic risk scores: Genomes to risk prediction. Clin. Chem., 69: 551-557.
- Steyerberg, E.W. and Y. Vergouwe, 2014. Towards better clinical prediction models: Seven steps for development and an ABCD for validation. Eur. Heart J., 35: 1925-1931.
- Harrell Jr., F.E., K.L. Lee and D.B. Mark, 1996. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat. Med., 15: 361-387.
- Uno, H., T. Cai, M.J. Pencina, R.B. D'Agostino and L.J. Wei, 2011. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat. Med., 30: 1105-1117.
- Blanche, P., J.F. Dartigues and H. Jacqmin-Gadda, 2013. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med., 32: 5381-5397.
- Rotimi, C.N., A.R. Bentley, A.P. Doumatey, G. Chen, D. Shriner and A. Adeyemo, 2017. The genomic landscape of African populations in health and disease. Hum. Mol. Genet., 26: R225-R236.
- Ogunniyi, T.J., B.S. Fatokun, K.O. Isah, A. Abdulbaki, A.R. Emiola and K. Batisani, 2025. Current status of cancer diagnosis and treatment in Nigeria. Health Sci. Rep., 8.
- Ngene, S.O., B. Adedokun, P. Adejumo and O. Olopade, 2018. Breast cancer genetics knowledge and testing intentions among Nigerian professional women. J. Genet. Couns., 27: 863-873.
- Collins, G.S., J.B. Reitsma, D.G. Altman and K.G.M. Moons, 2015. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): The TRIPOD statement. Ann. Intern. Med., 162: 55-63.
- Chung, C.C., E.C.Y. Su, J.H. Chen, Y.T. Chen and C.Y. Kuo, 2023. XGBoost-based simple three-item model accurately predicts outcomes of acute ischemic stroke. Diagnostics, 13.
- Buniello, A., J.A.L. MacArthur, M. Cerezo, L.W. Harris and J. Hayhurst et al., 2019. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res., 47: D1005-D1012.
|
|
How to Cite this paper?
APA-7 Style
Sowunmi,
K.S., Ogundele,
O.E. (2026). Polygenic Risk Score as a Predictor of Mammary Carcinogenesis in NMU-Treated Mice. Trends in Biological Sciences, 2(1), 74-86. https://doi.org/10.21124/tbs.2026.74.86
ACS Style
Sowunmi,
K.S.; Ogundele,
O.E. Polygenic Risk Score as a Predictor of Mammary Carcinogenesis in NMU-Treated Mice. Trends Biol. Sci 2026, 2, 74-86. https://doi.org/10.21124/tbs.2026.74.86
AMA Style
Sowunmi
KS, Ogundele
OE. Polygenic Risk Score as a Predictor of Mammary Carcinogenesis in NMU-Treated Mice. Trends in Biological Sciences. 2026; 2(1): 74-86. https://doi.org/10.21124/tbs.2026.74.86
Chicago/Turabian Style
Sowunmi, Kehinde, Sherifdeen, and Olusegun Emmanuel Ogundele.
2026. "Polygenic Risk Score as a Predictor of Mammary Carcinogenesis in NMU-Treated Mice" Trends in Biological Sciences 2, no. 1: 74-86. https://doi.org/10.21124/tbs.2026.74.86

This work is licensed under a Creative Commons Attribution 4.0 International License.